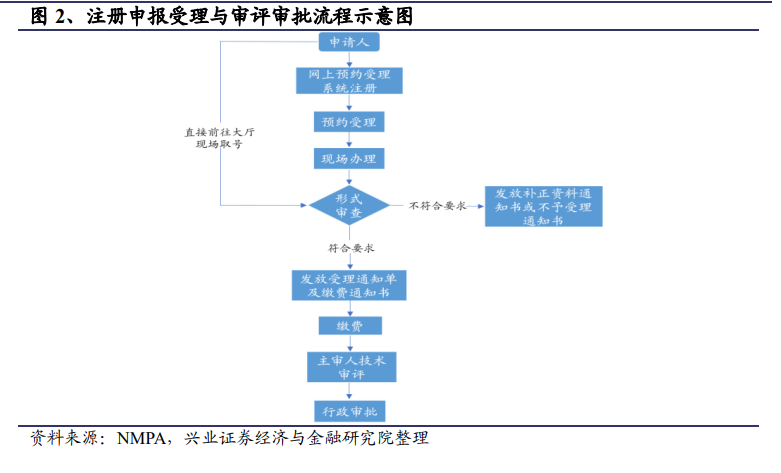
一般來說,第一類醫療器械實施備案管理,第二、三類醫療器械則實行注冊制管理,獲批注冊后方可上市銷售。具體的管理政策方面,國產第一類器械向設區的市級藥監部門提交資料即可;國產第二類器械由所在地省、自治區、直轄市的藥監部門進行審評審批;國產第三類和所有的進口產品則需要向國家藥品監督管理局申報。屬于創新、優先或藥械組合的產品在辦理進入相應流程后,可隨即進行產品類別判定。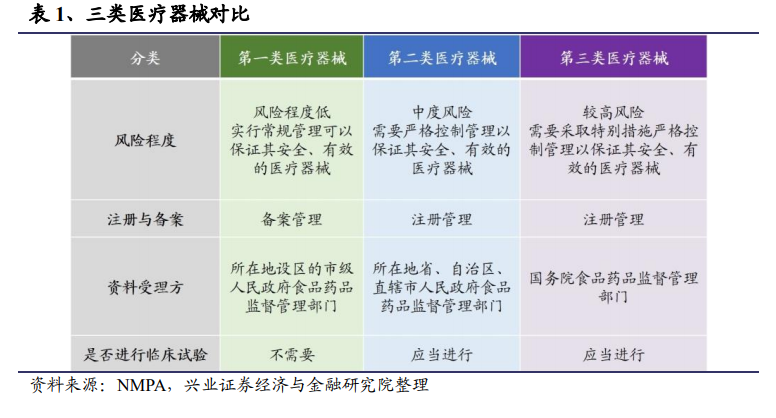
根據 2014 年 10 月 1 日起施行的《醫療器械注冊管理辦法》第十六條,申請第二類、第三類醫療器械注冊,應當進行注冊檢驗。醫療器械檢驗機構應當依據產品技術要求對相關產品進行注冊檢驗。第一類醫療器械實行備案管理,因此無需進行注冊檢驗,但備案人可以提交產品自檢報告。
醫療器械的注冊檢驗準備資料包括:符合醫療器械質量管理相關要求生產的樣品、產品技術要求、產品相關的技術資料等。檢驗工作的流程為:申請人與檢測中心簽訂檢驗合同、申請人提交產品技術要求及產品技術資料,將待檢驗樣品送至檢測中心、檢測中心開展檢測工作、檢測中心出具檢驗報告。