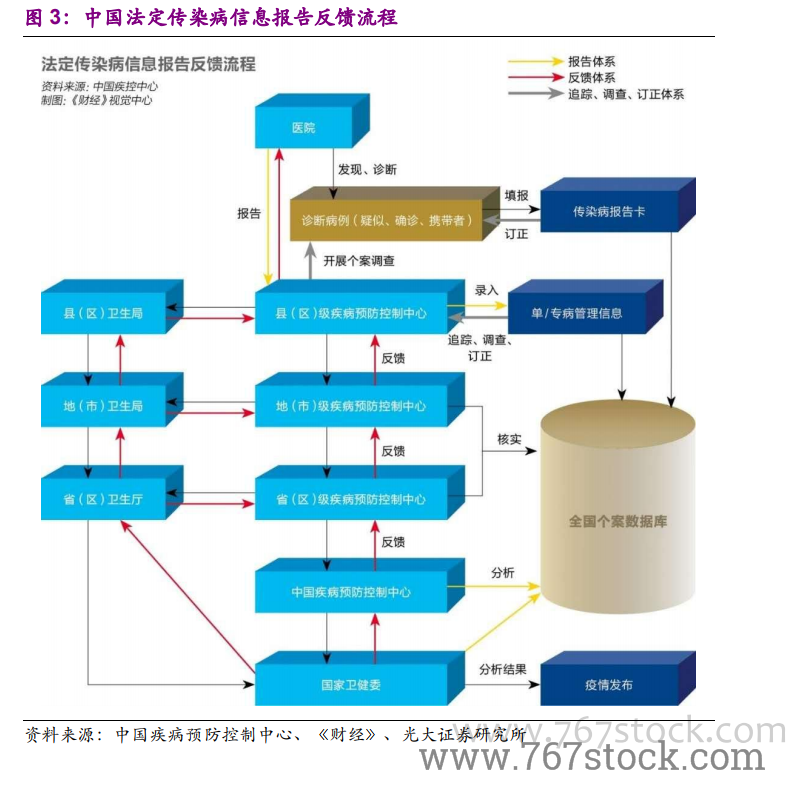
SARS 之前,新藥監管體系有待完善。在 SARS 之前,我國的藥審就處在大變革之中。1998年,為順應醫藥分離的改革需求,國家藥品監督管理局(SDA)成立,我國藥品監管開始走向規范。2003 年之前,SDA 剛剛建立了(藥品生產質量管理規范)GMP、(藥品經營質量管理規范)GSP 制度、生物制品批簽發制度、地標轉國標等工作,即剛剛完成了藥品生產、流通監管體系的搭建,以及藥品審批標準的全國統一。但在藥品研發的過程監管和特殊審批等方面則幾乎是空白。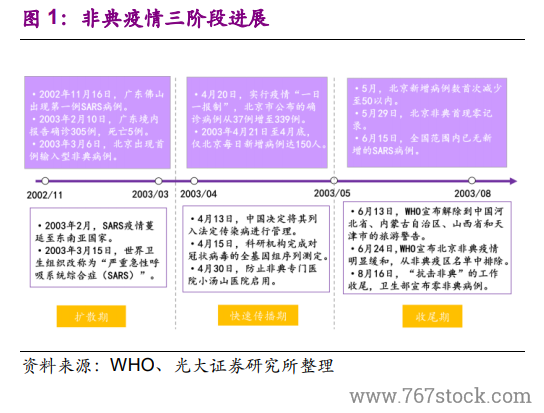
SARS 之下,“藥品加快上市注冊”程序應運而生。面對疫情,2003 年 4月 21 日,SFDA 印發《關于加強預防診斷治療非典型肺炎藥品和醫療器械監督管理的緊急通知》,建立預防、診斷、治療“非典”藥物快速審批通道,標志著我國藥審“藥品加快上市注冊”程序的正式建立。疫情期間,重組人干擾素α-2b 噴霧劑、連花清瘟膠囊、注射用烏司他丁等多個品種基于快速審批通道審批進入臨床研究。2005 年 11 月,SFDA 印發《國家食品藥品監督管理局藥品特別審批程序》,針對突發公共衛生事件的特別審批程序實現了制度化。此后,“藥品加快上市注冊”程序逐步完善,特殊審批程序、優先審評審批程序分別于 2007 年、2016 年建立。值得注意的是,2019 年 12 月10 日的《藥品注冊管理辦法(征求意見稿)》中提出了“突破性治療藥物”和“附條件批準程序”兩種程序。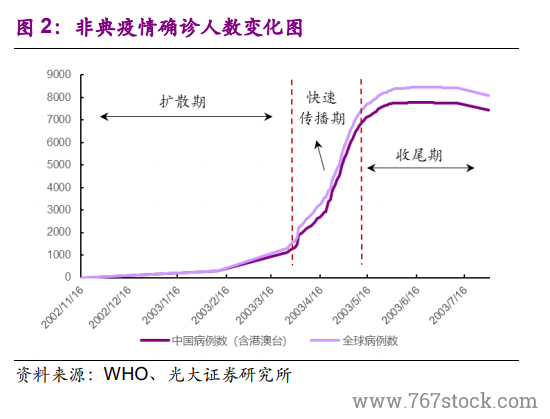
后 SARS 時代,GLP、GCP 制度建立。SARS 疫情之后,SFDA 繼續強勢推進藥審領域的改革。2003 年 8 月,SFDA 印發了《藥物非臨床研究質量管理規范》、《藥物臨床試驗質量管理規范》,建立了(藥物非臨床研究質量管理規范)GLP、(藥物臨床試驗質量管理規范)GCP 制度。從 1998 年到 2003 年,短短 5 年時間,SFDA 就在我國建立了 GMP、GSP、GLP、GCP、國家局統一藥審等現代化的醫藥監管制度。但在后期執行中,暴露出了腐敗在內的一系列問題,導致了 2007 年 SFDA 的降格,劃歸衛生部管理。此后,我國藥審領域的改革進度明顯放緩,直到 2013 年,SFDA 升格為 CFDA(部級),藥審改革又重回快車道。